Output Validation¶
DisCVR allows the validation of the classification output using the reference-assembly technique to assess significance of matches. During the build of the virus k-mer database in DisCVR, a library that contains the complete reference genomes of some of the viruses, which are represented by k-mers in the database, is generated. The tool allows two validations using the extracted reference genome library:
- k-mer Assembly: maps all classified k-mers to a reference genome.
2. Read Assembly: maps all sequence reads to a reference genome using Tanoti (a BLAST-guided aligner).
The validation takes place from the Full Analysis panel when the user right-clicks on the virus, which they wish to validate. A list with two items: k-mer Assembly and Read Assembly appears and once the user makes a selection, the reference-assembly starts. When the alignment is finished, a line graph showing the coverage and depth of the sequence data in the sample is displayed. A full coverage of the reference genome indicates strong evidence for the presence of the virus in the sample. The graphs can be saved as a PNG file for future reference. Figure 3 shows an example of the validation stage and its outputs.
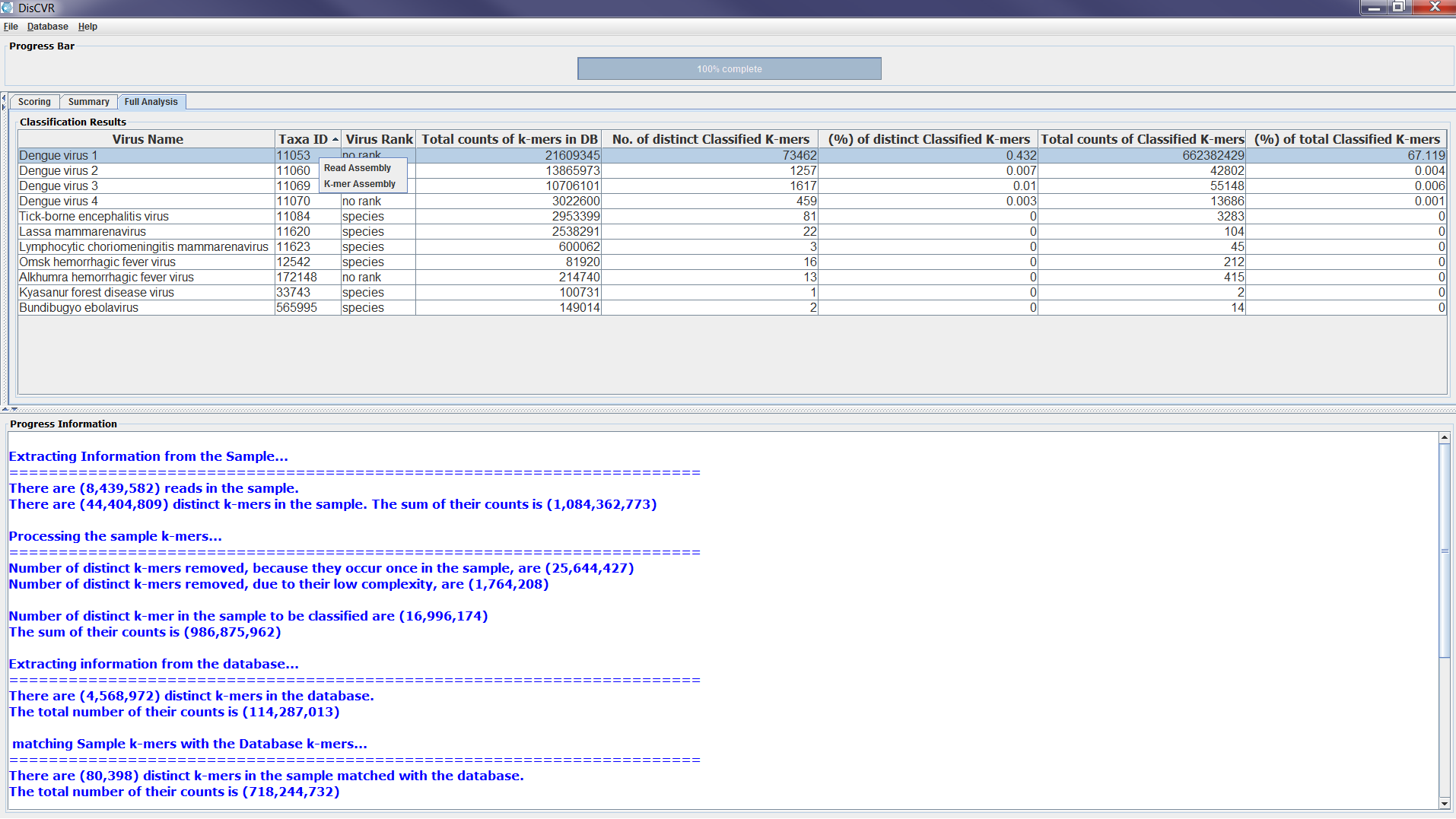
Figure4a: Screenshots showing how to assess the significance of matches from the sample classification results. Right-clicking on a row from the Full Analysis table provides a pop-up list of choices to start reference assembly
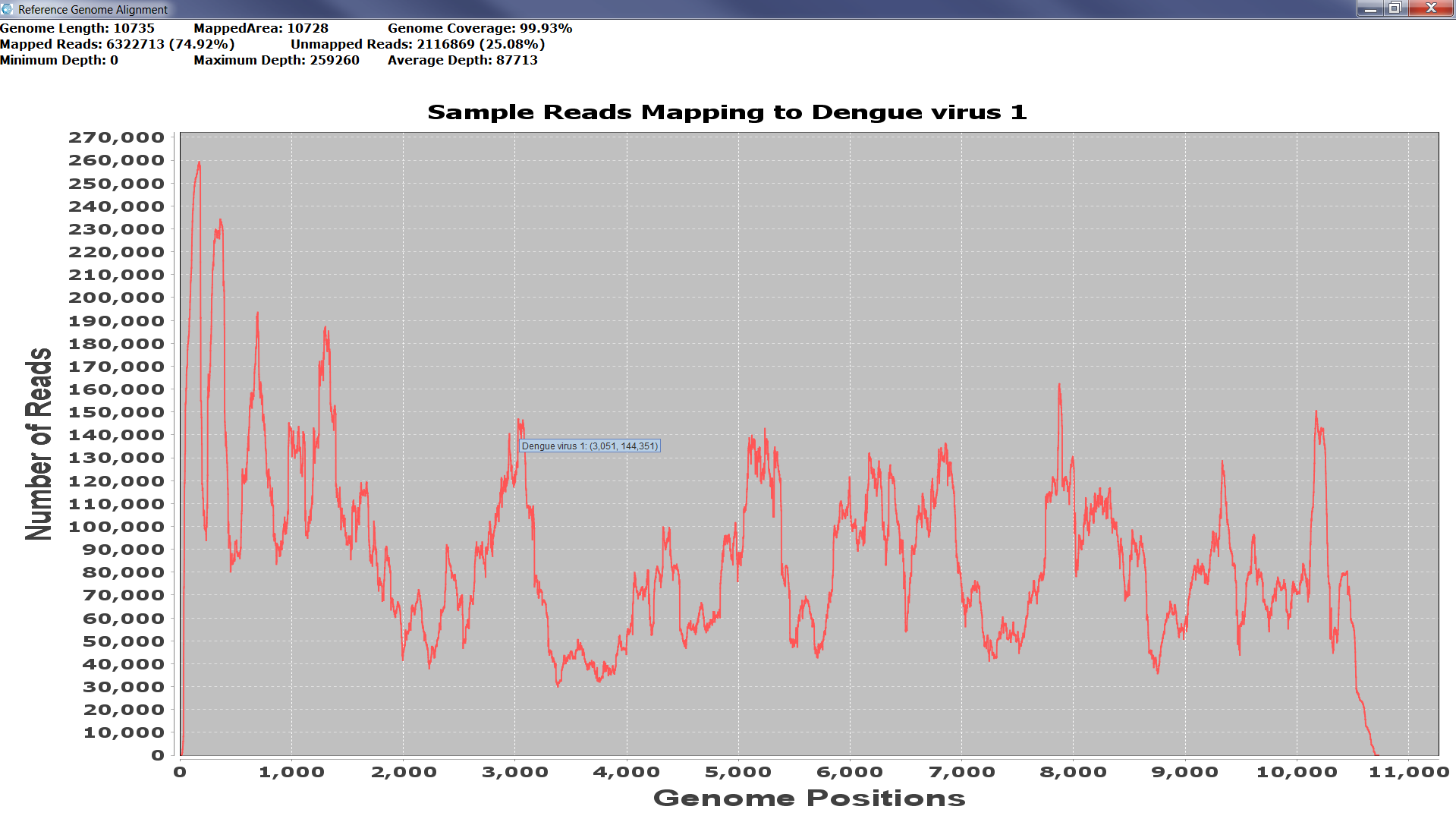
Figure4b: Screenshots showing how to assess the significance of matches from the sample classification results. Right-clicking on a graph e.g. Read-Assembly graph shows the number of reads at a certain reference genome position.
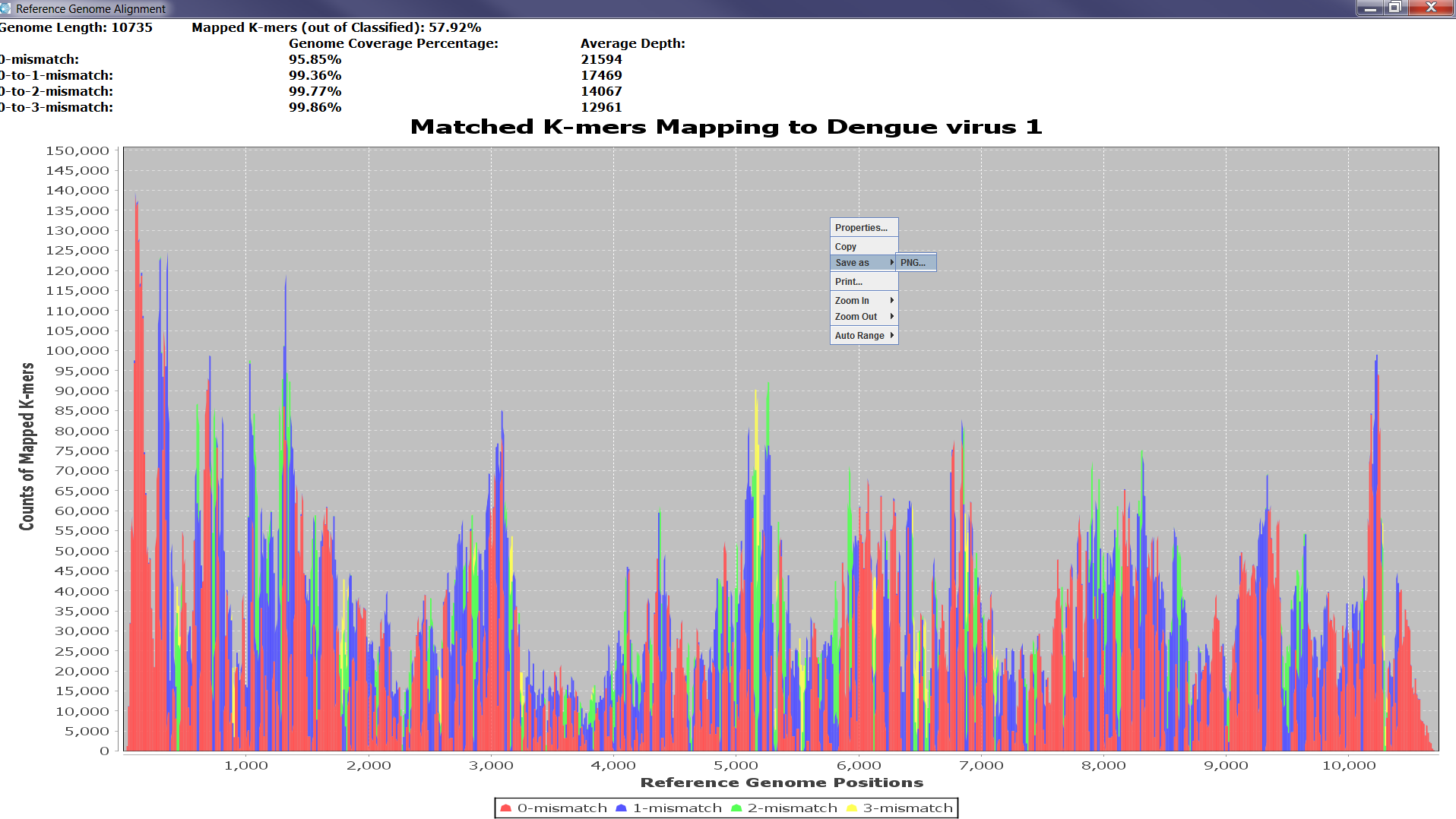
Figure4c: Screenshots showing how to assess the significance of matches from the sample classification results. Right-clicking on a graph e.g. k-mer assembly allows the users to save it as a PNG file.
DisCVR Command Line¶
DisCVR can be used to classify multiple samples at once using the command line. In this case, a folder is generated containing the results for each sample in a separate .csv file. The results consist of information about the reads in the sample and the full analysis of matched k-mers. The jar called DisCVR_CL.jar is used to run classifications on more than one file. Refer to the Installation section to download and install the jar and ensure that it is on the same path as DisCVR.jar and the lib folder. The following command can be used to run DisCVR_CL.jar to classify multiple files.
java –jar full/path/to/DisCVR_CL.jar <samples folder> <k> <file format> <Database name> <database option> <entropy threshold>
<samples folder>: is the full path to the folder which contains sample files to be classified. <k>: is the k-mer size. If using the DisCVR built-in database, use 22 <file format>: is the format of the sample files e.g. fastq <database name>: is the name of the database to be used in the classification. If DisCVR’s built-in database is to be used then use HaemorrhagicVirusDB_22, RespiratoryVirusDB_22, or HSEVirusDB_22. In case of a customisedDB, the full path to the database file must be provided. <database option>: if it is one of DisCVR’s database then use BuiltInDB and if it is a customised database file then use customisedDB. <entropy threshold>: is the threshold to use to remove low- entropy k-mers. If using DisCVR’s built-in database, for consistency use 2.5.
Using the command line, DisCVR enables the analysis of multiple clinical samples, however validation of the results via reference genome alignment is not available at this stage.
Building custom databases¶
- If you want to build a customised database, the following NCBI tools and files must be
downloaded and installed:
- The NCBI eutilities tools are used to download data. The tools can be found on NCBI eutlities. The full path to the edirect folder should be added to the system variables
- The NCBI taxdump files are used for taxonomy information retrieval when building a customised database. The file can be downloaded from NCBI taxonomy. The file taxdump.tar.gz should be downloaded and unzipped. The two files: names.dmp and nodes.dmp MUST be copied to the customised database folder: customisedDB which is in the same path as DisCVR.jar.
- To see if eutilities tools is added to the path:
esearchThis should state``Must supply -db database on command line```
Customised Database¶
DisCVR allows the users to build their own customised database from a list of viruses that are of interest. This section explains the steps to generate the users customised database files. Refer to the Installation section to ensure the required tools are downloaded and installed properly before proceeding to customise your k-mers database. The NCBI files, i.e. names.dmp and nodes.dmp, must be copied to the customisedDB folder. The NCBI website ([https://www.ncbi.nlm.nih.gov](https://www.ncbi.nlm.nih.gov)) is used for downloading the data. The following table lists all the files and parameters needed to build a customised database.
| Files/Parameters | Description |
|---|---|
| Input File | A file containing information about the set of viruses to build the database from |
| NCBI Taxdump Files | Two files (names.dmp and nodes.dmp) to be downloaded into the customisedDB folder |
| Host genomes file | A fasta file containing the host DNA sequences |
| Entropy threshold | A number in the range [0,3] to act as a low-complexity threshold |
| Data Location | The path to the folder containing the data to build the database from |
| Name of the database | The given name to the customised database. This should be a single word that does NOT contain (_) |
| K size | The length of k-mers to be used in the build of the database. |
| Number of threads | The number of threads to use during the build of the database. |
| File counter | The number of virus files to process at one time during the build of the database. |
Table 1: Files and parameters needed to build DisCVR’s customised database
The process starts by providing a list of the viruses of interest and the information of their complete genomes, if they exist. This input is a tab-delimited file which contains the taxonomy ID of the virus and the accession number for its reference sequence. In DisCVR, the three built-in databases use only human viral sequences at the species and subspecies levels on the taxonomy tree. However, the customised-database does not require the viruses to be of a particular rank in the taxonomy tree nor the host to be human. In addition, the input file should not have duplicate taxonomy IDs but multiple taxonomy IDs in the list can have the same reference sequence. Table 2 shows an example of the input file.
IMPORTANT: the accession number of the virus reference sequence MUST be the exact accession number provided in the header of the reference sequence.
There are three steps to build the customised database which must be executed sequentially:
- Data Download; which involves obtaining the data required to build the two database files.
- Generating the reference genome library file using the information of the reference sequences.
- Generating the virus k-mers database from the downloaded sequences.
| Taxid | Accession Number | Description |
|---|---|---|
| 121791 | NC_002728.1 | The virus taxonomy ID is followed by the accession number of the virus’s reference sequence. |
| 499556 | NC_010563.1,NC_010562.1 | It is a segmented virus. The accession numbers for the reference sequences segments are comma-separated and listed in order so that the first segment is the largest. |
| 11598 | Only the virus taxonomy ID is provided because the virus does not have a reference sequence. |
Table 2: An example of the input file for the customised database builds stage. Left column shows an example of a line in the input file. Right column explains the components of the line.
Data Download¶
The first step in building the database is to obtain the viral sequences for the viruses of interest. The script downloadDataAndRefSeq.sh uses the eutilities tools to download the data from the NCBI using the following command:
bash downloadDataAndRefSeq.sh <taxIDs_List> <outputDir>
<taxIDs_List>: the input file which contains the virus taxonomy IDs and their reference sequence information. <outputDir>: the name of the directory for the downloaded data.
After executing the script, the output directory will have two sub-directories:
- DataSeq which contains the data to be used to build the virus k-mers file, and
- RefSeq which contains the data to create the reference sequence library file.
Each file in both directories includes in its name the taxonomy ID (taxID) of the virus whose data is contained in the file. The DataSeq directory has two corresponding files for each taxID:
- Virus_taxID.fa is a fasta file which contains all complete and partial viral sequences for the virus with the taxID, and
- Virus_taxID_Info which contains information for the sequences in the corresponding fasta file. The first line states the number of sequences found on NCBI for the taxID followed by information for each sequence. This includes Accession number, title, gi header, update date, length of the sequence, the subtype, and strain.
Similarly, the RefSeq directory contains files for each taxID which has information of its reference sequence in the input file. The files which are called Virus_taxID_RefSeq.fa are fasta files that contain one or more sequences (in the case of segmented viruses). Information files are used to identify segmented viruses, in the case of RefSeq, and to filter out shared sequences, in the case of DataSeq.
IMPORTANT: ensure that the files in both RefSeq and DataSeq folders are NOT empty. If any of the file are empty, re-run the data download script with an input file that contains only the information for the taxIDs with empty files.
Database Files Generation¶
Java programs are used to generate the two files in the database. These Java programs are found in the folder called ‘bin’.
Reference Genome Library¶
The reference genome library file is used in the validation stage of DisCVR and it contains the reference sequences for the viruses in the input file. The reference sequences are identified in the input file by their accession numbers and the corresponding sequences are downloaded in the RefSeq directory. The Java program GenomesLibrary is used to generate the reference genome library using the following command:
java –cp ./bin customdatabase.GenomesLibrary <taxIDs_List> <database_Name> <RefSeq_Dir>
<taxIDs_List>: the input file which contains the virus taxonomy IDs and their reference sequence information. <database_Name>: is the name to be given to the customised database < RefSeq_Dir >: is the full path of the RefSeq folder which contains all downloaded data for the reference sequences.
The output file is called: databaseName_referenceGenomesLibrary and it is in the RefSeq folder. Each line in the file consists of a virus taxonomy ID, the header, and the sequence of its complete genome. The delimiter “@” is used to separate the three components. In the case where the reference sequence is a concatenation of multiple segments, the headers are separated by a comma and the sequences are separated by a sequence, 300 in length, of the letter N. For the viruses which share the same reference sequence (i.e. species level and their sublevels are assigned the same reference sequence) then their taxonomy IDs are separated with a pipe sign “|”.
The output file MUST be copied to the customisedDB folder to be used for DisCVR’s validation when using the customised k-mers database.
Virus k-mers database¶
The last step to build the customised database is to generate the file which contains the virus k-mers. In the generation of DisCVR’s built-in database files, a filtering step was used to remove shared sequences between a virus and its ancestors in the taxonomy tree. This is an optional step which can be used in the build of the customised database to reduce redundancy in the data and to increase virus specificity.
Data Filtering (Optional)¶
The filtering process removes shared sequences from the ancestors’ data. For example, if sequences are found in both the strain and the species levels, then they are removed from the sequences at the species level. It is recommended to keep a copy of the data in the DataSeq directory before executing this step to avoid loss of data. The NCBI dump files (i.e. names.dmp and nodes.dmp) are used in this step and they MUST be included in the customisedDB folder. The Java program DataSequences is used to filter the downloaded sequences using the following commands:
mkdir <DataSeq_filtered>
cp -r <DataSeq_Dir> <DataSeq_filtered>
java –cp ./bin customdatabase.DataSequences <DataSeq_Dir> <genomes_file> <database_Name>
< DataSeq_Dir>: is the full path for the DataSeq folder which contains the viruses’ data (fasta and Info files). <DataSeq_filtered>: is the full path for the folder which contains the filtered data. <genomes_file>: is the full path for the referenceGenomeLibrary which is generated from the previous step. <database_Name>: is the name to be given to the customised database.
After executing the program, the sequence files (i.e. fasta files) for the viruses which are ranked at a higher taxonomic level to other sequences in the DataSeq directory are modified, if they contain shared sequences. The output file: databaseName_lineageID.txt shows, per line, the taxID of a virus followed by the taxIDs of higher taxonomic level. In addition, a file which contains summary information about the viruses in the database such as their rank in the taxonomy tree and the number of sequences per virus, after filtering, is generated. The file is called: databaseName_DataInformation.csv.
Finally, an output file called: databaseName_allSeqData is generated and it contains all the sequences, after filtering, from all the data files in the directory. The three output files can be found in the DataSeq directory.
k-mers database file¶
The final step in the customised database build is to generate the virus k-mers database file from the downloaded viral sequences. In this step, k-mers from the viral sequences and from the host genomes are extracted. Low entropy and host k-mers are then removed from the virus k-mers. The remaining set of virus k-mers are assigned the taxIDs of all the viruses which are represented by these k-mers. Table 3 shows an example of the virus k-mer database file.
| k-mer | count | taxids |
|---|---|---|
| AAAAACAAGAATGGACAC | 2 | 11620 1 |
| AAAAACAATGGGCTCTAT | 5 | 1980486 1980491 2 |
Table 3: An example of the virus k-mers database file. Each line contains a k-mer of size 18, the number of times it occurs in the viral sequence, and a list of taxIDs of the viruses which the k-mer is extracted from. The last number in the line indicates whether the k-mer occurs in a single virus (e.g. 1) or in multiple related viruses (e.g. 2).
The virus k-mers database file is generated by using the following command and it is saved to the customisedDB folder.
java –cp ./bin customdatabase.KmersDatabaseBuild
<DataSeq_Dir> <host_file> <kmersDB_file> <k_size> <num_threads> <entropy_threshold> <files_counter> <DataSeq_Dir>: full path to the directory which contains a copy of the downloaded data (fasta files). <host_file>: full path to the host genomes fasta file. <kmersDB_file>: name to be given to the customised k-mers database file. <k_size>: k-mer size. <num_threads>: number of threads to use when counting k-mers from the virus files. We recommend using 2 <entropy_threshld>: entropy threshold to filter out low-entropy k-mers. Use 0 to omit this option. <files_counter>: number of data files to process at once during taxonomy labelling. It is recommended to use 10 to optimise memory use.
An example¶
This section shows an example of the whole process to create the customised k-mers database files. The NCBI dump files: names.dmp and nodes.dmp must be downloaded from the NCBI into the customisedDB folder before starting the process. In addition, the lib folder must be in the same directory as the customisedDB folder. The input files provided in the TestData are used in this example. There are two input files:
- miniDB_sample.txt: contains information of the viruses used to build the k-mers database.
- HumanGenomesTest.fa: contains an example of the host sequences
The process starts by downloading the data from the NCBI and generating the reference genome file. Filtering of shared sequences is then applied on a copy of the viral sequences. The last step is creating the virus k-mers database with k size equals to 18 and 0 entropy threshold. The number of threads to count k-mers is 2 and the number of files to process at once is 10. The given name for the customised database files in this example is “miniDB_sample”.
bash downloadDataAndRefSeq.sh ./TestData/miniDB_sample.txt ./TestData/miniDB_sample/
mkdir ./TestData/miniDB_sample/DataSeq_filtered
cp -r ./TestData/miniDB_sample/DataSeq/* ./TestData/miniDB_sample/DataSeq_filtered
java -cp ./bin customdatabase.GenomesLibrary ./TestData/miniDB_sample.txt miniDB ./TestData/miniDB_sample/RefSeq/
cp ./TestData/miniDB_sample/RefSeq/miniDB_referenceGenomesLibrary ./customisedDB/
java -cp ./bin customdatabase.DataSequences ./TestData/miniDB_sample/DataSeq_filtered ./TestData/miniDB_sample/RefSeq/miniDB_referenceGenomesLibrary miniDB
java -cp ./bin customdatabase.KmersDatabaseBuild ./TestData/miniDB_sample/DataSeq_filtered/ ./TestData/HumanGenomesTest.fa miniDB_Kmers_18 18 2 0 10