Welcome to VATK’s documentation!
¶
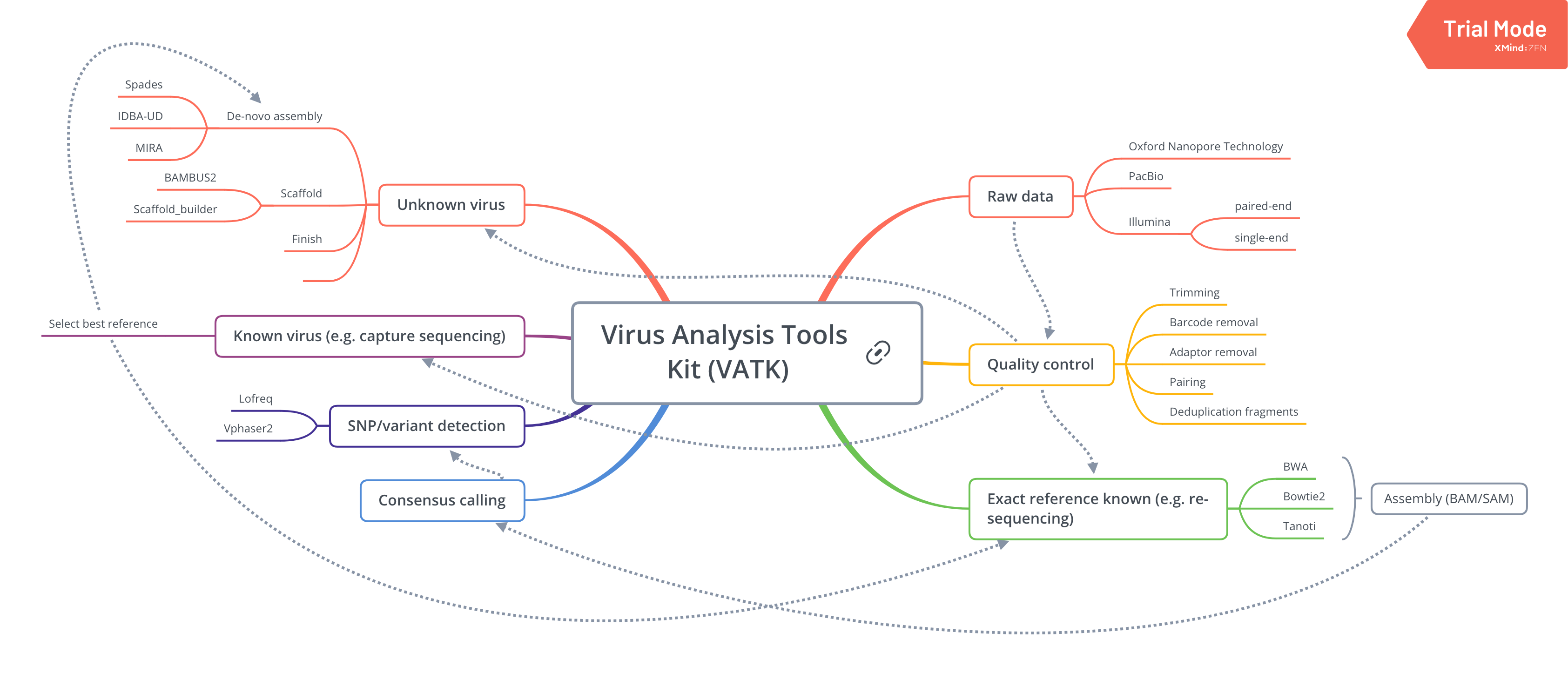
Contents:
Evaluation of sequence data
Counting sequences
Checking sequence quality
Complexity of the sequencing data
Trimming
Tools
How to run
Benchmark
References
Pre-assembly duplicate removal
Reference selection
Genotype detection
Reference selection
De-novo assembly
Alignment/mapping
BWA
bowtie2
Tanoti
Post-alignment analysis
Detecting chimeras
Coverage smoothing
Further contamination removal
Counting and removing duplicates
Realignment around indels
Removing reads with indels
Remove the duplicate fragments
Reporting alignment stats
Depth of coverage
Breadth of coverage
Standard error and covariance
weeSAM
Pipelines
HCMV pipeline
Indices and tables
¶
Index
Module Index
Search Page
VATK
Navigation
Contents:
Evaluation of sequence data
Trimming
Pre-assembly duplicate removal
Reference selection
Alignment/mapping
Post-alignment analysis
Reporting alignment stats
Pipelines
Related Topics
Documentation overview
Next:
Evaluation of sequence data
Quick search